

FLUORESCENT IN SITU HYBRIDIZATION ANALYSIS
OF CHROMOSOMAL MICRO-DUPLICATIONS AND
MICRO-DELETIONS IN CLINICAL GENETICS
Sarri C*
*Corresponding Author: Dr. Catherine Sarri, Genetics Department, Institute of Child Health, in Aghia Sophia Childrens Hospital, Thivon and M. Asias, Goudi, 115 27 Athens, Greece; Tel: +3 0 210 7467789; Fax: +3 0 210 7700111; E-mail: inchildh@otenet.gr
page: 73
|
|
SYNDROMES OF MICRO-DUPLICATIONS
Charcot-Marie-Tooth type 1A (CMT1A). It is the first delineated micro-duplication syndrome and is associated with a micro-duplication of chromosome 17 (region 17p11.2) which contains PMP22, an important gene for peripheral nervemyelination. The main symptom of the syndrome is sensorimotor neuropathy [3,4]. Inheritance is autosomal dominant in pattern. Analysis of nerve biopsies suggests that the disorder is caused by increased gene dosage. Occasionally, CMT1A results from point mutations within the PMP22 gene. In cases with a duplication, onset of symptoms is usually in the first decade of life, while slowing of nerve conduction velocity is evident from the age of 2 years. The most common clinical phenotype is the CMT syndrome with distal muscle wasting and weakness, tendon areflexia, usually mild sensory loss, and foot deformity.
Smith-Magenis. Ninety percent of patients with Smith-Magenis syndrome have a micro-deletion of chromosome 17 (region 17q11.2), while a few cases (about 10%) have been described with micro-duplication of the same region. The main features of a micro-deletion are: mental retardation, congenital anomalies and behavioral problems, while a micro-duplication has a milder phenotype [5,6].
Beckwith-Wiedemann. The cardinal features of this disorder, which is most often referred to as Beckwith-Wiedemann syndrome (BWS), are exophthalmus, macroglossia and gigantism in the neonate. These patients are at risk of developing specific tumors. Beckwith-Wiedemann syndrome is caused by a mutation in the chromosome 11p15.5 region. Duplication in this region appears to be involved in the pathogenesis, and imprinting results in anomalous patterns of transmission. Most cases are sporadic [7,8].
Downs Syndrome with Normal Karyotypes. Downs syndrome with a normal karyotype is caused by a micro-duplication of chromosome 21 (region 21q21-21q22.1) not seen even with high resolution conventional cytogenetic techniques. The main features of this well recognized syndrome are: mental retardation, characteristic facies, heart defects, hypotonia, simian crease, etc. [9,10].
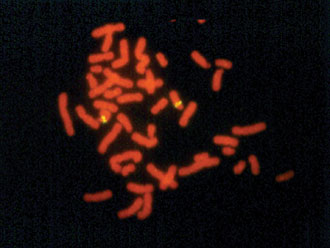
Figure 1. Williams syndrome with a micro-deletion.
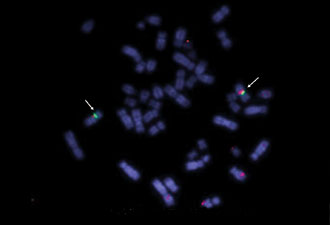
Figure 2. Di-George syndrome with a micro-deletion.
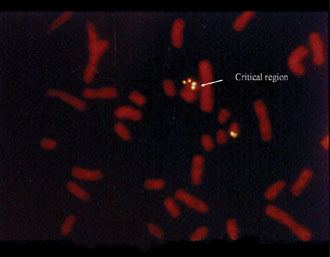
Figure 3. Prader-Willi region without a micro-deletion.
|
|
|
|

 |
Number 28
Vol 28 2025 Supplement |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|
|