

PRE-IMPLANTATION GENETIC DIAGNOSIS FOR
β-THALASSEMIA, SICKLE CELL SYNDROMES
AND CYSTIC FIBROSIS IN GREECE
Traeger-Synodinos J1,*, Vrettou C1, Tzetis M1, Palmer G2,
Davis S3, Mastrominas M3, Kokali G4, Pandos K4, Kanavakis E1
*Corresponding Author: : Dr. Joanne Traeger-Synodinos, Medical Genetics, Athens University, Choremio Research Laboratory, St. Sophias Childrens Hospital, Thivon and Levadias Streets, Athens 11527, Greece; Tel.: +30-210-746-7461; Fax: +30-210-779-5553; E-mail: jtraeger@cc.uoa.gr
page: 25
|
|
PRE-IMPLANTATION GENETIC DIAGNOSIS FOR β-THALASSEMIA AND SICK¬LE CELL SYNDROMES
The thalassemia syndromes and related hemoglobinopathies are the commonest group of monogenic autosomal recessive disorders world-wide [10]. They are caused by mutations in the β-globin gene located on chromosome 11. More than 170 point mutations or small insertions/deletions have been described which either reduce or abolish the synthesis of the β-globin chains of adult hemoglobin (Hb A) by the affected gene (http://globin.cse.psu. edu/globin).
When confronting a common and molecularly heterogeneous monogenic disease such as the β hemoglobinopathies, it is more practical to have a single PGD diagnostic strategy applicable for a wide spectrum of potential affected genotypes, rather than designing and standardizing case-specific protocols each time. Several methods applicable to PGD of hemoglobinopathies have been described for the detection of some of the numerous β gene mutations [11-14], but only a few have been described for a potentially wider application [15,16]. The initial protocol we developed was based on nested PCR and denaturing gradient gel electrophoresis (DGGE), and exploited the observation that the majority of the common β-thal mutations in most populations worldwide tend to be clustered within the first 700 nucleotides of the β-globin gene [17,18]. Denaturing gradient gel electrophoresis is advantageous for application to PGD, since as a scanning method, it can identify any mutation within a single amplified region, precluding the need for independent mutation assays, and additionally, it facilitates simultaneous analysis of more than one mutation within a single PCR fragment, invaluable when confronting compound genotypes. In addition, DGGE is an assay for the presence of normal as well as pathological alleles, and in practice, only blastomeres with definitive evidence of a normal allele on DGGE analysis are considered unaffected, preventing transfer of affected embryos, even if ADO has occurred.
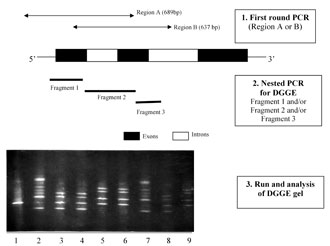
The diagnostic strategy involved a first PCR to amplify a several hundred base pair (bp) region, followed by nested PCR to generate amplicons suitable for DGGE analysis (Fig. 1). The pre-clinical experiments were carried out on 490 single cells (blastomeres from supernumerary human embryos, lymphocytes and amniocytes), through which the genotyping method was optimized to achieve a PCR efficiency of 85-90%, with less than 8% allele drop-out [17,18].
Between 1998 and 2002, 59 couples at risk for transmitting β hemoglobinopathies were counseled about PGD. Forty-one couples initiated 63 PGD cycles (one cycle in 22 couples, two cycles in 16 couples, and three cycles in three couples), of which 20 cycles with <4 IVF embryos were canceled before biopsy and genotype analysis. Amongst 43 completed cycles, 302 cleavage stage embryos were biopsied, 236 (78%) gave a genotype result, of which 125 clearly had at least one normal allele with DGGE analysis (unaffected). Transfer of 100 embryos (1-4/cycle) established 16 pregnancies (37% for each completed cycle), including 13 singletons, two sets of twins and one set of triplets (20% implantation per embryo transferred). Six pregnancies were lost within the first trimester but 10 underwent second trimester PND. Nine pregnancies (13 babies) were confirmed unaffected, but one singleton was a PGD misdiagnosis, and was selectively terminated. The misdiagnosis was attributed to extraneous contamination or a tube switch, and not to inaccuracy of the genotyping method. The triplet pregnancy was selectively reduced to twins, and all pregnancies went to term, resulting in the birth of 12 healthy babies [19].
Although accurate, the DGGE-based protocol is technically demanding, time consuming, and not as sensitive as methods involving fluorescent PCR. To simplify PGD analysis, reduce diagnosis time, improve sensitivity, whilst maintaining accuracy and monitoring of ADO, we recently established a protocol based on real-time PCR, using fluorescent hybridization probes for mutation detection. We designed, standardized and validated mutation detection probes for the common β-thal mutations worldwide (and Hb S), through mutation analysis in >200 carriers, and additionally, 25 prenatal diagnoses. We adapted the method to PGD using nested PCR, with the second reaction in LightcyclerÔ capillaries (Roche Diagnostics GmbH, Mannheim, Germany), including fluorescent detection probes for melting curve analysis and allele assignment. So far, we have applied the LightcyclerÔ (Roche Diagnostics) protocol in 10 PGD cycles. Results (available within 5 hours) were obtained in 81/89 blastomeres (91%), of which 69 blastomeres were also analyzed with the DGGE-PGD protocol, and genotypes were completely concordant. Thus, PGD using mutation detection with real-time PCR is accurate, but additionally, it is more sensitive and rapid, when compared to the DGGE-based method [20,21].
Figure 1. The PGD strategy for b-globin gene mutations. 1) First round PCR (region A or B); 2) nested PCR for DGGE fragment 1 and/or fragment 2 and/or fragment 3; 3) run and analysis of the DGGE gel. The DGGE gel stained with ethidium bromide. Lane 1: normal; lane 2: control sample, compound heterozygote for mutations IVS-I-110 (G®A) and IVS-I-6 (T®C); lanes 3-7: single blastomeres from five embryos; lane 8: parent, heterozygote for IVS-I-110; lane 9: parent, heterozygote for IVS-I-6.
|
|
|
|

 |
Number 28
Vol 28 2025 Supplement |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|
|