

GENE ALTERATIONS LEADING TO HYPOXANTHINE-
GUANINEPHOSPHORIBOSYL TRANSFERASE
DEFICIENCY: GENOTYPE-PHENOTYPE CORRELATION
Neychev VK*, Krastev SR, Mitev VI
*Corresponding Author: Dr. Vladimir K. Neychev: Department of Chemistry and Biochemistry, Medical University, 2 Zdrave str., Sofia 1431, Bulgaria; Tel: +359-889-49-51-25 (personal), +359-2-51-66-528 (office); Fax: +3592-952-02-07; E-mail: Neychev@dir.bg
page: 51
|
|
DISCUSSION
We have attempted to relate the different sites or types of reviewed mutations with particular clinical variants of HPRT deficiency. Such a correlation would be important in the prediction of disease severity for cases identified in the neonatal period or by prenatal screening. Unfortunately, we found no precise correlation of the location or nature of the available mutations with particular clinical variants.
However, the majority of cases with less severe clinical variants 1 and 2 have point mutations, leading to amino acid substitution (see Table 1), rather than insertions, deletions, nonsense mutations or splice site errors. An amino acid substitution is less likely to disrupt the enzyme function than other types of mutations. Indeed, point mutations that are associated with the less severe clinical variants 1 and 2, predicted more than 1.5% of residual enzyme activity, while those associated with clinical variants 3 and 4 predicted less than 1.5% of residual activity. Thus, identification of such a mutation with concurrent measurement of residual enzyme activity could permit reasonable prediction of clinical variants.
The majority of deletions, insertions, nonsense and splice site mutations resulted in clinical variant 4. However, the few cases, resulting in less severe clinical variants (see Tables 2-5), appear to be an exception to the concept that the disease severity depends on the value of residual activity of the enzyme [1], since they led to complete HPRT deficiency.
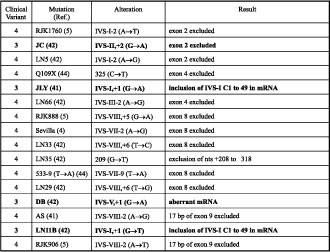
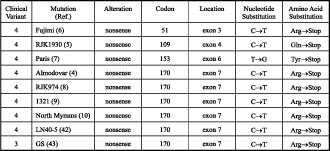
The nonsense mutation HPRT GS was reported as clinical variant 3 because of the lack of self-mutilation by age 10 [6] (see Table 3). Exactly the same mutation but called HPRT (Almodovar) [7], HPRT (RJK974) [8], HPRT (1321) [9], HPRT (North Mymms) [10] and HPRT (LN40-5) [5] resulted in clinical variant 4. This discrepancy could be due to the possible appearance of the self-mutilation at a later age, since this could delay until as late as 18 years of age [1]. One would expect that patients with HPRT GS would develop clinical variant 4 at a later age. It is most likely that some of the patients with clinical variant 3 will never develop typical LNS features because of their shortened lifespan [1].
Two deletions, HPRT (Illinois) and HPRT (Japan 3) (see Table 4), and one insertion, HPRT RW (see Table 5), are other examples for the above-mentioned exception. The HPRT (Illinois) is a deletion that spans nts 12 to +1, and result in the juxtaposition of a G to the remaining UG of the initiation codon. It has been proposed that the residual activity may be a result of inefficient translational initiation at the newly formed, unusual GUG initiation codon [11]. The HPRT (Japan 3) [12] is a 51 bp deletion in exon 9 that spans nts +648 to +698, and produced HPRT protein missing only the last two amino acids. The HPRT (RW) is a 3 bp addition at nt 429 that resulted in the introduction of an additional amino acid without disturbing the normal reading frame [3].
Despite the fact that the majority of splice site mutations produced clinical variant 4 the prediction of phenotypic outcome may be difficult, since four of these resulted in the less severe clinical variant 3 (see Table 2). These are HPRT (JLY) [4], HPRT (LN11B) [5], HPRT (JC) [5] and HPRT (DB) [5], representing IVS-I,+1 (G®A), IVS-I,+1 (G®T), IVS-II,+1 (G®A) and IVS-V,+1 (G®A), respectively. Perhaps these mutations, somehow producing a portion of correctly spliced transcripts, could result in small amount of functional enzyme.
There are several examples of mutations, which implies that the location of the mutations has less significant influence on phenotypic outcome (see Table 1). In HPRT (Madrid I) the replacement of glycine by valine at codon 71 caused clinical variant 1, while the replacement by arginine [HPRT (Yale)] caused clinical variant 4. Similarly, the replacement of arginine by proline [HPRT (Banbury)], glycine [HPRT (Toronto)] and glutamine [HPRT (Tsou)] at codon 51 caused clinical variants 4, 1 and 2, respectively.
There are many cases in which the same mutation resulted in the same clinical variants in different unrelated patients. One would expect that some prediction of the clinical variant of a newly found case could be possible, finding a mutation similar to those that were already found. However, there are a few cases with the same mutation that have produced quite different clinical variants, even in the same family. For example, four male siblings from the family with HPRT (Moos Jaw) displayed quite different clinical variants (two had a learning disability, one has a speech impediment and one was neurologically normal) [13]. Here the expression of the different clinical variants could be influenced by factors other than molecular mutation. This observation implies that even identification of identical mutation makes the prediction of disease severity of a newly found case quite uncertain.
Table 1. Inherited missense mutations, leading to the hypoxanthine-guanine phosphoribosy transferase deficiency.
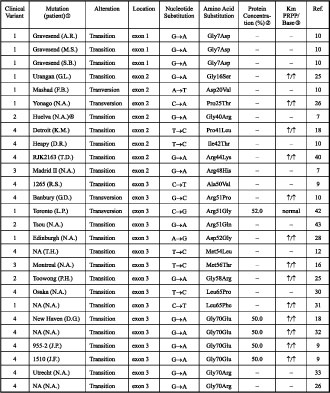
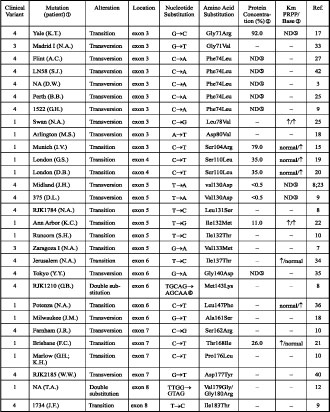
Note: the protein size was normal in all cases.
The name of the mutations is that assigned by the group defining the mutation. A place name such as Osaka indicates that the mutant protein has been called HPRTOSAKA; RJK+numbers are codes used by the Robert J. Kleberg, Jr., Center for Human Genetics, Baylor College of Medicine, Houston, TX, USA; and GM numbers are codes used by the National Institute of General Medical Sciences Human Genetic Mutant Cell Repository.
The intracellular concentration is given as a percentage of normal levels, where this assay was employed.
ƒ Up arrows indicate an increase in Km value.
No data available.
HPRT activity not detected.
Two nts are altered; the second one does not lead to an amino acid substitution.
Table 2. Mutations resulting in splicing errors.
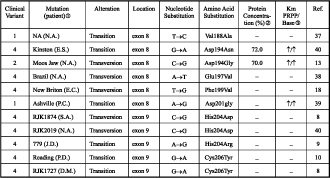
Table 3. Mutation resulting in a premature stop codon.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|