

THE UNSTABLE HEMOGLOBINS:
SOME GENETIC ASPECTS
Wajcman H1,*, Galacteros F2
*Corresponding Author: Dr. Henri Wajcman, INSERM U 468 and Service de Biochimie, Hôpital Henri Mondor AP-HP, 51 Avenue du Maréchal de Lattre de Tassigny, 94010 Créteil, France
page: 3
|
|
UNSTABLE HEMOGLOBINS CARRIED BY THE β GENE
The variants affecting the β gene were the first unstable Hbs to be described. From a biological and clinical point of view, they form a very heterogeneous group. The most classical situation is that in which the variant leads to a protein present in the peripheral blood at a level lower than that of Hb A. This Hb is more sensitive than Hb A to oxidant stress in the peripheral blood, causing its precipitation as Heinz bodies on the membrane of the cell. The rigidified and fragile erythrocytes are pitted and eliminated by the spleen, leading typically to a chronic hemolytic anemia exacerbated by crises consecutive to infections or oxidant drugs. Hemoglobins, which precipitate in the bone marrow in the minutes following their biosynthesis, form another group of unstable variants: they lead to an anemia with some degree of ineffective erythropoiesis similar to that observed in thalassemic syndromes.
Each of the mutations responsible for an unstable Hb affects only a limited number of individuals in a few families, usually unrelated. De novo mutations are frequently observed since these mutations are always a cause of disease. They do not bring any selective advantage and are, thus, eliminated in the forthcoming generations. In industrialized countries the founder effects are usually identified, since any patient suffering from a hemolytic anemia will be investigated until the cause of his/her disease is explained.
For reasons which are still unclear, Hb Köln [β98 (FG5)Val→Met] is the most frequently encountered unstable variant [3]. It has been described in various European countries (France, Spain, Italy, Czechoslovakia, Russia, etc.) and in several populations from Asia (China, Japan, Korea, etc.) where it was also known as Hb Ube 1 [6-11]. It is frequently observed in families where several members are affected, but many de novo cases have also been reported. In the simple heterozygous state it leads to a moderately severe hemolytic anemia. A compound heterozygous case was reported in which it was associated with a β0-thal, and resulted in a thalassemia intermedia phenotype [12]. The main abnormal functional property of Hb Köln is to lose some heme groups from the β chains [13]. Therefore, in RBC, or in a fresh hemolysate, about 15-20% of Hb Köln forms semi-Hb, a molecular species detected by electrophoresis, which displays a high oxygen affinity and stimulates erythropoietin secretion and erythropoiesis. As a consequence, the Hb level is only moderately decreased but this is associated with some degree of impaired oxygen binding transport, causing functional anemia.
The numerous cases of Hb Köln described worldwide, as well as the finding of the same mutation in Hb A2-Wrens [14], a d chain variant, and in Hb Medecine Lake [15], a globin chain carrying two mutations, suggest that codon 98 could be a mutation hot-spot.
In contrast, very few patients, or even only a single one, have been reported for each of the unstable variants causing really severe hemolytic anemia. In the classic review of Nute and Stamatoyannopoulos [16], on 90 Hb variants produced by de novo mutations, 63 were unstable. Frequently the abnormality affects a single individual in a family and arose through a de novo mutation. In other cases, the parents have no abnormal Hb but several children may be affected, either because they are monozygous twins [17] or as an effect of a germinal mosaic [18].
It is likely that the high severity of the anemia and its complications hinders the diffusion of these mutations, making the finding of familial cases exceptional, and increasing the frequency of de novo cases in the published literature. In our experience, the heterozygous carriers of Hbs Bicźtre [b63(E7)His→Pro], Saint Etienne [β92(F8) His→Gln], Saint Louis [β28(B10)Leu→Gln], and Hammersmith [β42(CD1)Phe→Ser], who were followed in France, had their anemia partly controlled by splenectomy, but these patients died from thromboembolic complications, being young adults without children.
Hb Hammersmith is a severely unstable variant [4,19-21]. In this Hb the CD corner is modified, there is no heme loss but the structural change of the heme pocket leads to low oxygen affinity and high auto-oxidation rate, with rapid formation of highly unstable hemichromes [22]. The Hb level is always lower than in Hb Köln because of the decreased oxygen affinity, and it may drop considerably when submitted to oxidative stress. To our knowledge, all the described cases of Hb Hammersmith concern de novo mutations. Thus, some correlation exists, probably between the frequency of an unstable variant and the severity of the disorder it leads to. In the case of Hb Bicźtre, about half of the circulating red cells were reticulocytes, indicating that the life span of the RBCs was approximately 2 days [23], and only two cases have been reported in the literature [24].
The finding of several genetic markers, such as microsatellites in the parents chromosomes, demonstrates that the mutation results from a de novo event. In the case of Hb Duino [β92(F8)His→Pro;β104(G6)Arg→Ser], the de novo mutation is obvious. In this Italian family, several members carried Hb Camperdown [β104(G6)Arg→Ser] but one presented with a severe hemolytic anemia: its abnormal β chain carried the mutation of Hb Camperdown and that of Hb Newcastle [β92(F8)His→Pro], known to be a severely unstable variant [25] (Fig. 1).
The phenotype of an unstable Hb may be modified by epistatic factors that could be genetic or environmental. For example, an unstable Hb may be better tolerated when associated with a high level of Hb F, resulting from a hereditary persistence of Hb F. Carbon monoxide (CO) is known to stabilize Hb Zürich [β63(E7)His→Arg], which is thus better tolerated by smokers than by non smokers, as a result of the 65-fold increase in the affinity of the abnormal β-globin chain of this variant for this ligand [26]. Conversely, hereditary hyperbilirubinemias, found in the Gilbert syndrome, increase the risk for gall bladder lithiasis, and mutations such as the factor V Leiden mutation or some mutations in the prothrombin gene may facilitate thromboembolic complications. Interaction with other Hb variants is always possible: Hb Saki [β14(A11)Leu→Pro] was found to be associated to a β-thal trait leading to thalassemia intermedia [27], and in another observation to Hb S, with a phenotype expressing a higher level of Hb S than Hb Saki and resulting in a hemolytic sickle cell syndrome [28].
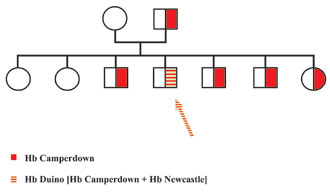
Figure 1. Pedigree of family with Hb Duino. In this family several members including the father carried Hb Camperdown [b104(G6)Arg®Ser], a very slightly unstable variant causing no hematological symptoms. In one of the siblings who suffered from a marked hemolytic anemia, the Hb study showed that the abnormal chain of this patient carried, together with the mutation of Hb Camperdown, a second mutation identified as being that of Hb Newcastle [b92(F8)His®Pro], another mutation known to cause instability and hemolytic anemia.
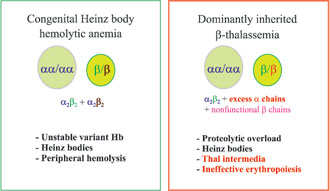
Figure 2. Schematic differences between the two categories of unstable b chain variants.
Figure 3. Electrospray ionization mass spectrometry analysis of the hemolysate from the carrier of Hb Esch. This patient had borderline a+-thalassemic RBC parameters, and IEF revealed a faint band between the positions of Hb F and Hb A. The ESI-MS showed a small signal, 400 mass units above the normal value for a chains, which corresponded to no known adduct but rather to some complex variant. This Hb was found, by DNA sequencing, to result from a duplication of the sequence Ala65-Leu-Thr-Asn68, which is in good agreement with the difference in mass.
|
|
|
|

 |
Number 27
VOL. 27 (2), 2024 |
Number 27
VOL. 27 (1), 2024 |
Number 26
Number 26 VOL. 26(2), 2023 All in one |
Number 26
VOL. 26(2), 2023 |
Number 26
VOL. 26, 2023 Supplement |
Number 26
VOL. 26(1), 2023 |
Number 25
VOL. 25(2), 2022 |
Number 25
VOL. 25 (1), 2022 |
Number 24
VOL. 24(2), 2021 |
Number 24
VOL. 24(1), 2021 |
Number 23
VOL. 23(2), 2020 |
Number 22
VOL. 22(2), 2019 |
Number 22
VOL. 22(1), 2019 |
Number 22
VOL. 22, 2019 Supplement |
Number 21
VOL. 21(2), 2018 |
Number 21
VOL. 21 (1), 2018 |
Number 21
VOL. 21, 2018 Supplement |
Number 20
VOL. 20 (2), 2017 |
Number 20
VOL. 20 (1), 2017 |
Number 19
VOL. 19 (2), 2016 |
Number 19
VOL. 19 (1), 2016 |
Number 18
VOL. 18 (2), 2015 |
Number 18
VOL. 18 (1), 2015 |
Number 17
VOL. 17 (2), 2014 |
Number 17
VOL. 17 (1), 2014 |
Number 16
VOL. 16 (2), 2013 |
Number 16
VOL. 16 (1), 2013 |
Number 15
VOL. 15 (2), 2012 |
Number 15
VOL. 15, 2012 Supplement |
Number 15
Vol. 15 (1), 2012 |
Number 14
14 - Vol. 14 (2), 2011 |
Number 14
The 9th Balkan Congress of Medical Genetics |
Number 14
14 - Vol. 14 (1), 2011 |
Number 13
Vol. 13 (2), 2010 |
Number 13
Vol.13 (1), 2010 |
Number 12
Vol.12 (2), 2009 |
Number 12
Vol.12 (1), 2009 |
Number 11
Vol.11 (2),2008 |
Number 11
Vol.11 (1),2008 |
Number 10
Vol.10 (2), 2007 |
Number 10
10 (1),2007 |
Number 9
1&2, 2006 |
Number 9
3&4, 2006 |
Number 8
1&2, 2005 |
Number 8
3&4, 2004 |
Number 7
1&2, 2004 |
Number 6
3&4, 2003 |
Number 6
1&2, 2003 |
Number 5
3&4, 2002 |
Number 5
1&2, 2002 |
Number 4
Vol.3 (4), 2000 |
Number 4
Vol.2 (4), 1999 |
Number 4
Vol.1 (4), 1998 |
Number 4
3&4, 2001 |
Number 4
1&2, 2001 |
Number 3
Vol.3 (3), 2000 |
Number 3
Vol.2 (3), 1999 |
Number 3
Vol.1 (3), 1998 |
Number 2
Vol.3(2), 2000 |
Number 2
Vol.1 (2), 1998 |
Number 2
Vol.2 (2), 1999 |
Number 1
Vol.3 (1), 2000 |
Number 1
Vol.2 (1), 1999 |
Number 1
Vol.1 (1), 1998 |
|
|